










研究課題
量子力学の発見以来その化学への適用は大きな成功を収め、今日では化学の基本原理として量子化学の地位は確立されました。有機化学の分野においても福井謙一先生のフロンティア軌道理論、ウッドワード・ホフマン則等、適用範囲の広い理論が生まれました。当研究室では分子軌道理論、密度汎関数理論、軌道相互作用理論その他の手法を用いて、分子の構造、物性、反応性等に関する幅広い研究を行っています。最近では計算機の性能が著しく向上し、大規模な現実系の理論計算が精度良く行えるようになっており、化学の広い分野で有効に使われています。以下の研究課題が我々の主なターゲットです。
分子デバイスに関する理論的研究
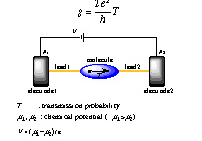
現在のシリコンベースのコンピューターシステムに取って代わるものとして、単一分子を利用したデバイスに注目が集まっている。1997年、Tourらのグループによってベンゼンジチオール単一分子に流れる電流を測定した実験は非常によく知られたものである。我々は単一分子デバイスのコンダクタンスを計算するための方法としてランダウアモデル (図1) を採用し、分子軌道法に立脚した計算プログラムを独自に開発し研究を行っている。計算対象としてグラファイトを用いた研究では、コンダクタンスと分子軌道との重要な相関関係の導出に成功し、分子デバイスとして有効な分子、接続方法などを提案している。その他の計算対象として、ベンゼンジチオール、ポルフィリン、DNA (図2) などがある。
 |
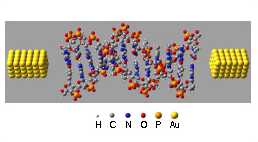 |
| 図1. ランダウアモデルによる量子細線のコンダクタンスq。 | 図2. (AT)12DNA分子の金と導線からなる分子ジャンクション。 |
分子性超伝導に関する理論的研究
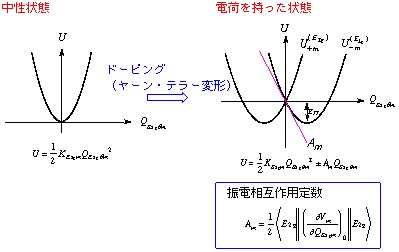
最近、常識を打破るような超伝導性が多くの分子性固体に発現することが見い出されている。高い対称性を持つ分子は縮退した分子軌道を持つが、この縮退した軌道に部分的に電子が詰まると、分子構造は不安定になり一定の変形が起こる (ヤーン・テラー効果)。そのような例としてフラーレン、大環状炭素、縮合芳香族炭素等が挙げられる。金属や合金などの特長と考えられていた超伝導性が、ごく一般的な有機化合物にも普通に見られることが明らかになっている。これらの分子材料における電子状態と分子振動のカップリング機構 (これを振電相互作用という) を解明し、その特異な超伝導性の解明を目指して研究を行っている。
生体化学反応に関する理論的研究
生体を維持制御しているのはさまざまな酵素であり、多くの場合その活性中心には鉄、銅、マンガン等の遷移金属が存在している。それら金属は困難な化学反応を生理的な条件下で繰り返し失敗なく行っている。例としてチトクロム P450やメタンモノオキシゲナーゼといった酸化酵素が挙げられるが、これらはアルカン類を容易にアルコールに変換する。メタン等の不活性アルカンの化学修飾は現代化学の目指す最重要課題のひとつであり、これらの反応機構が解明されれば、高性能人工触媒の開発も夢ではない。金属酵素のメカニズムを探るためには不安定中間体や遷移状態の情報を得ることが不可欠であるが、それらを実験から探ることは困難なことも多い。そこで本研究室では古典トラジェクトリー計算やQM/MM法といった量子化学からのアプローチにより生体化学反応の解明に新たな展開を計っている。
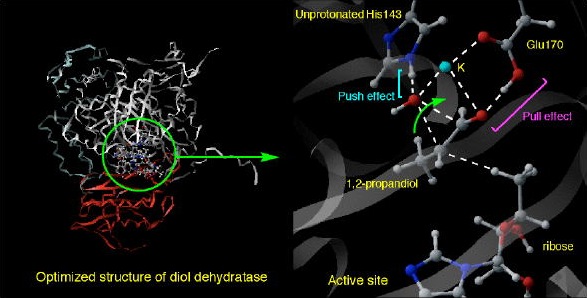 |
| ジオールデヒドラターゼ酵素の全原子計算 (15000原子) |
有機ケイ素化合物の構造と反応性に関する理論的研究
有機ケイ素化合物はさまざまな化学反応の出発物質あるいは反応中間体として有用である。ケイ素は炭素lsと同族でありながらその反応性は大きく異なっており、有機ケイ素化合物の反応において、Woodward-Hoffmann則のような軌道対称性の議論が成り立つのかどうかについてはまだよく分かっていない。有機ケイ素化合物の反応性を探るため、その反応中間体や遷移状態を密度汎関数法などの手法を使って理論的に解析している。
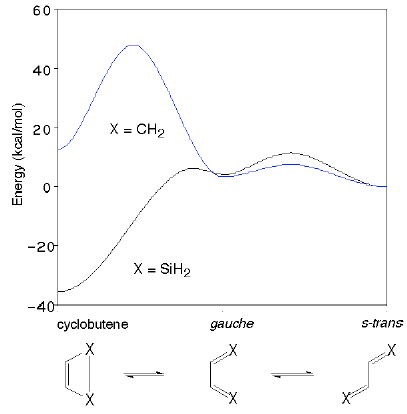 |
| シクロブテンとブタジエンの相対エネルギー |
接着に関する理論的研究
接着剤を用いた材料の接合は工業的に非常に重要な技術であり、自動車産業や航空産業をはじめとする多くの工業分野で利用されている。材料の接着性を向上させるために多くの研究がこれまでに行われているが、接着がどのような界面相互作用により起こるのかは十分には明らかにされておらず、古くから議論の対象となっている。特に原子レベルでの接着界面の解析は実験的研究では非常に困難であり、理論計算によるアプローチが期待されている。理論計算による接着機構の解析は今までほとんど行われていなかったが、近年の計算機や計算理論の進歩により表面や界面といった大規模系の計算が可能となった。本研究室では量子化学計算を用いて、接着相互作用に関する分子論的な研究を世界に先駆けて行っている。
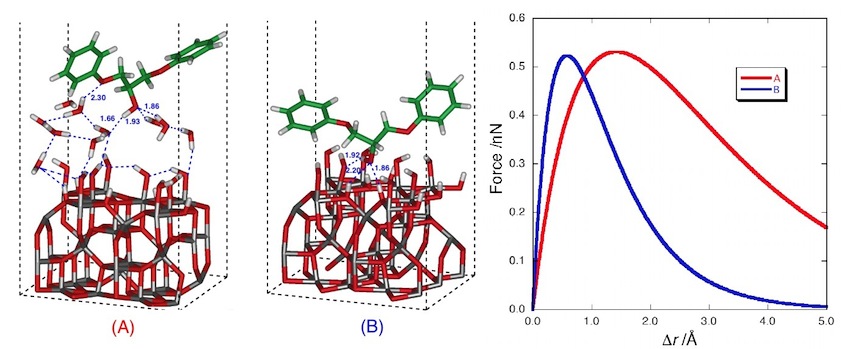 |
| アルミニウムーエポキシ樹脂界面の構造と計算で求めた接着力 |